
Li Li’s team Cooperation Findings: Histone Methyltransferase SETD2 regulates HCC occurence
November 04, 2020
Li Li’s team from School of Biomedical Engineering, Shanghai Jiaotong University, along with Min Wu’s team from School of Life Sciences, Wuhan University, published "Deficiency of stone methyltransferase SETD2 in liver leads to abnormal lipid metabolism and hepatocarcinoma" in the international famous journal Hepatology on October 15, 2020. It confirmed that SETD2 deficiency promotes the occurrence of hepatocellular carcinoma (HCC), and elucidates the mechanism of SETD2 regulating cholesterol homeostasis and c-Jun/AP1 signal transduction in hepatocellular carcinoma.
HCC is a common malignant tumor in the world. Many new cases occurred in Asia, especially in China. Liver plays an important role in cholesterol metabolism. The occurrence and development of HCC is a very complex biological process, which is systematically regulated by the body. HCC causes great damage to liver functions, leading to pathological changes in many metabolic processes including cholesterol metabolism. However, the regulatory mechanism of cholesterol metabolism in HCC development is still unclear.
Multiple enzymes exit in mammals for H3K36 methylation, including SET domain-containing 2 (SETD2), nuclear receptor-binding SET domain protein 1, Wolf-Hirschhorn syndrome candidate 1, Wolf-Hirschhorn syndrome candidate 1-like 1, PR domain 9, and absent, small, or homeotic-like. (1) Among these, SETD2 is the closest homologue of yeast Set2 and the major enzyme for H3K36me3 in mammalian cells. In 2014, associate researcher Li Li established a conditional gene knockout murine model of SETD2, and carried out extensive functional research in reproduction, development and cancer (Zuo et al, J bio Chem, 2018; Wang et al, PLoS Biol, 2018; Xu et al, Nat Genet, 2019; Ji et al, Nat Commun,2019; Niu et al,Gut, 2020). Based on this, using a liver-specific SETD2 depletion model, the two teams found that SETD2 deficiency is sufficient to trigger spontaneous HCC. Meanwhile, SETD2 depletion significantly increased tumor and tumor size of a diethylnitrosamine-induced HCC model.

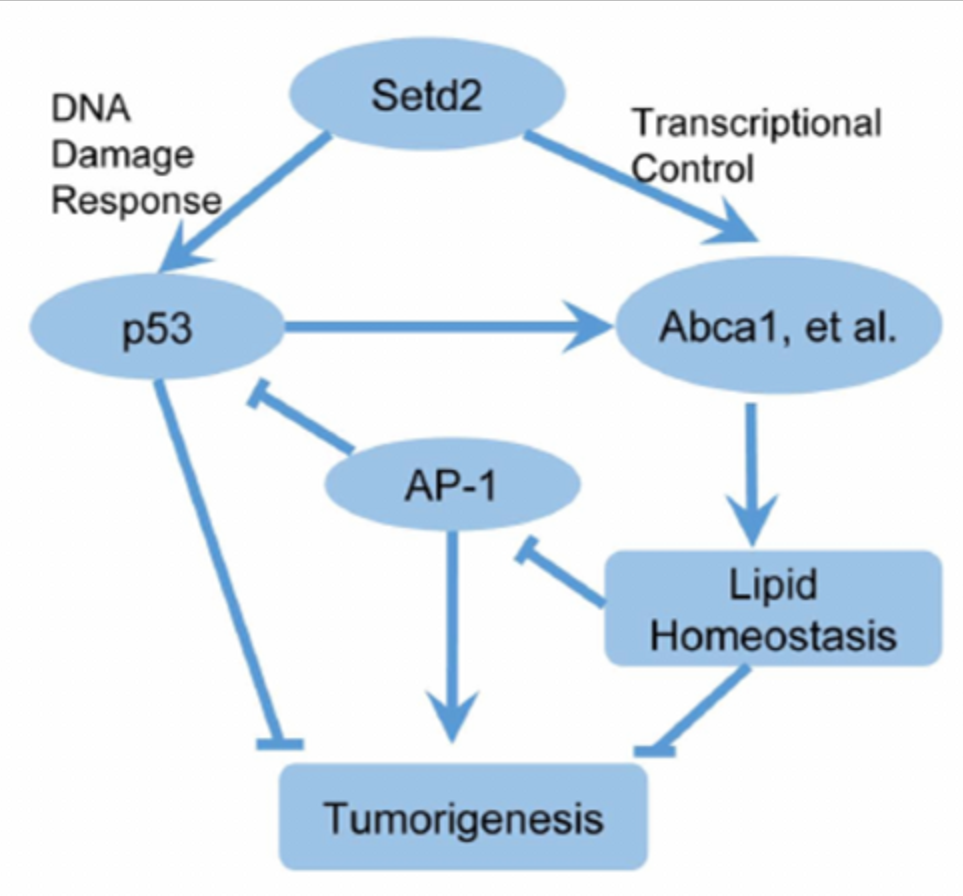
The mechanistic study showed that SETD2 suppresses HCC not only through modulating DNA damage response, but also by regulating lipid metabolism in the liver. SETD2 deficiency down-regulated H3K36me3 enrichment and expression of cholesterol efflux genes and caused lipid accumulation. High-fat diet enhanced lipid accumulation and promoted the development of HCC in SETD2-deficient mice. Chromatin immunoprecipitation sequencing analysis further revealed that SETD2 depletion induced c-Jun/activator protein 1 (AP-1) activation in the liver, which was trigged by accumulated lipid. c-Jun acts as an oncogene in HCC and functions through inhibiting p53 in SETD2-deficient cells.
This study was the first to reveal the role of SETD2 in HCC and mechanism of regulating cholesterol homeostasis and c-Jun/AP1 signal transduction, providing a new theoretical basis for HCC targeted therapy.
Xuejing Li from Wuhan University is the first author of this paper with Professor Wu and associate researcher Li Li as corresponding authors. The research project is supported by the Ministry of Science and Technology, National Natural Science Foundation of China and Shanghai Science and Technology Commission.
Full text link:
Address:Wenxuan Building, 800 Dongchuan RD, Minhang District, Shanghai, China
Tel: +86 21 34204018
Zip: 200241
Address:No3 Teaching Building, 1954 Huashan RD, Xuhui District, Shanghai, China
Tel: +86 21 62933344
Zip: 200030

BME WeChat
copyright: Copyright@2018 School of Biomedical Engineering, Shanghai Jiaotong University
Shanghai Jiaotong ICP Bei 05227
沪交ICP备20210179